血液腫瘤科/細胞治療中心/台灣細胞免疫醫學會 陳駿逸醫師
乳癌(Breast Cancer)在臨床上面臨異質性與多藥抗藥性(MDR)的雙重挑戰
在臨床上,我們經常會面對棘手的多藥抗藥性(Multidrug Resistance, MDR)乳癌。乳癌是一群高度異質性的腫瘤,傳統上依據雌激素受體(ER)、黃體素受體(PR)及人體表皮生長因子受體2(HER2)的表達分為三大主要臨床亞型:ER+(適用Tamoxifen等內分泌治療)、HER2+(適用Trastuzumab標靶治療) 以及三陰性乳癌(依賴Taxanes與Anthracyclines等化療與免疫檢查點抑制)。
然而,不論是先天性(de novo)或後天獲得的抗藥性,往往在病程進展中交織出現,高達30%的早期患者會面臨復發。要擊破這座堡壘,我們必須深入其底層的分子機制。
趨同與演變:抗藥性的多維問題背景
PI3K/Akt 訊息傳遞路徑、miRNA 調控以及表觀遺傳學突變,都是導致乳癌各亞型抗藥性交織的關鍵中心點。探究抗藥性的上游,雖然各亞型的第一線治療手段相異,但其走向抗藥的分子網絡卻表現出驚人的「趨同演變」:
- 內分泌治療抗藥性: 以 Tamoxifen 為例,除了 CYP2D6 的基因多態性影響產物 Endoxifen 的生物活性外,ERα 自身位點(如 :Serine 305)被 PKA 磷酸化,會使其結構構型改變,讓 Tamoxifen 由拮抗劑「黑化」成為激動劑。此外,變體 ER-α36 的表現增加,亦會讓其繞過配體依賴性的轉錄,直接激活下游路徑。
- Trastuzumab 治療抗藥性: 除了 Mucin 4 或 CD44/Hyaluronan 複合物造成的抗原表位掩蓋(Epitope Masking)阻礙標靶結合外,生成缺乏結合域、但具備本質性激酶活性的 p95-HER2 變體,也是常見的抗藥機制。
- 化療抗藥性:常因 MDR1(ABCB1)基因轉錄增加,導致 P-glycoprotein(P-gp)等 ATP 結合匣蛋白(ABC Transporters)高度表現,加速胞內化療藥物的泵出。
這些錯綜複雜的抗藥網絡,其背後其實共享了三大軸心機制:PI3K/Akt 訊息傳遞路徑的異常激活、microRNA(miRNA)的基因靜默調控,以及 DNA 甲基化與組蛋白修飾等表觀遺傳學改變(Epigenetic Alterations)。
分子剎車失靈的動態解密
核PI3K/Akt 路徑過度激活、miRNA-21/221 表現異常與 DNA 的異常甲基化,共同催化了腫瘤細胞的生存與增殖。

雌激素、HER2訊息傳遞路徑和PI3K/Akt路徑在抗藥性乳癌中的作用。
註:雌激素受體(ER)可透過核轉位活化基因轉錄,其活化方式包括配體結合後(1)或在沒有配體的情況下透過受體磷酸化(2)。 ER也可在SRC和其他銜接蛋白存在的情況下與質膜結合。此時,配體結合透過活化訊號路徑(包括PI3K/Akt和Ras/MAPK路徑(未顯示))觸發非基因組效應(3)。這些路徑也可被配體與GPR30結合(4)以及生長因子與受體酪胺酸激酶(包括HER2)結合激活,進而誘導自身磷酸化與下游訊號傳導(5)。如前所述,PI3K/Akt 路徑 (6) 是本文討論的三種乳癌抗藥性機制的匯聚點,因為路徑過度活躍經常伴隨多種下游效應 (7)。
如果把控制乳癌細胞增殖與凋亡的機制,比喻為一輛「正在下坡的高速賽車」,那麼 PI3K/Akt 路徑 就是這輛車的油門放大器。在正常狀況下,名為 PTEN 的抑癌蛋白是這輛車的主剎車系統,負責拉住油門,不讓細胞無限增殖。 然而在抗藥性乳癌中,發生了嚴重的「剎車失靈」,其過程有下列的情況:
- 油門被卡死(路徑突變): 由於 PI3K 催化亞基(p110α)發生突變或擴增,油門被直接踩到底,Akt 被源源不絕地磷酸化激活。
- 剎車皮被磨平(miRNA 介入): 腫瘤內高表現的 miR-21 就像是剪斷剎車線的黑手,它直接結合並降解 PTEN 的 mRNA,讓主剎車系統徹底癱瘓。
- 剎車踏板被焊死(表觀遺傳的鎖定): 透過 DNMT1 催化的 PTEN 啟動子高度甲基化(Hypermethylation),就像是用一把無形的「表觀遺傳鎖」把剎車踏板死死焊在底部,使其無法發揮作用。
當這套剎車系統全線崩潰,下游的下游效應器 mTORC1 就會瘋狂運轉。它不僅促使轉錄因子 FoxO3a 被磷酸化並排除於細胞核外(失去抑制細胞週期的能力),更進一步去激活抗凋亡蛋白(如: BCL-2、Survivin)的表現。此時,不論我們在外給予多少 Tamoxifen 或 Trastuzumab,這輛瘋狂的賽車都完全無視阻礙,載著腫瘤細胞一路狂奔,達成抗藥性與無限增殖。
此外,表觀遺傳的調控不僅鎖住了剎車,還同時啟動了逆向泵。在化療耐藥細胞中,MDR1 基因啟動子發生了低甲基化(Hypomethylation) 以及組蛋白 H3K9 的高度乙酰化,這等於是為癌細胞加裝了無數個高效能的排氣管(P-gp 泵),在化療藥物(如 :Taxanes)進到胞內發揮作用前,就將它們悉數排出。

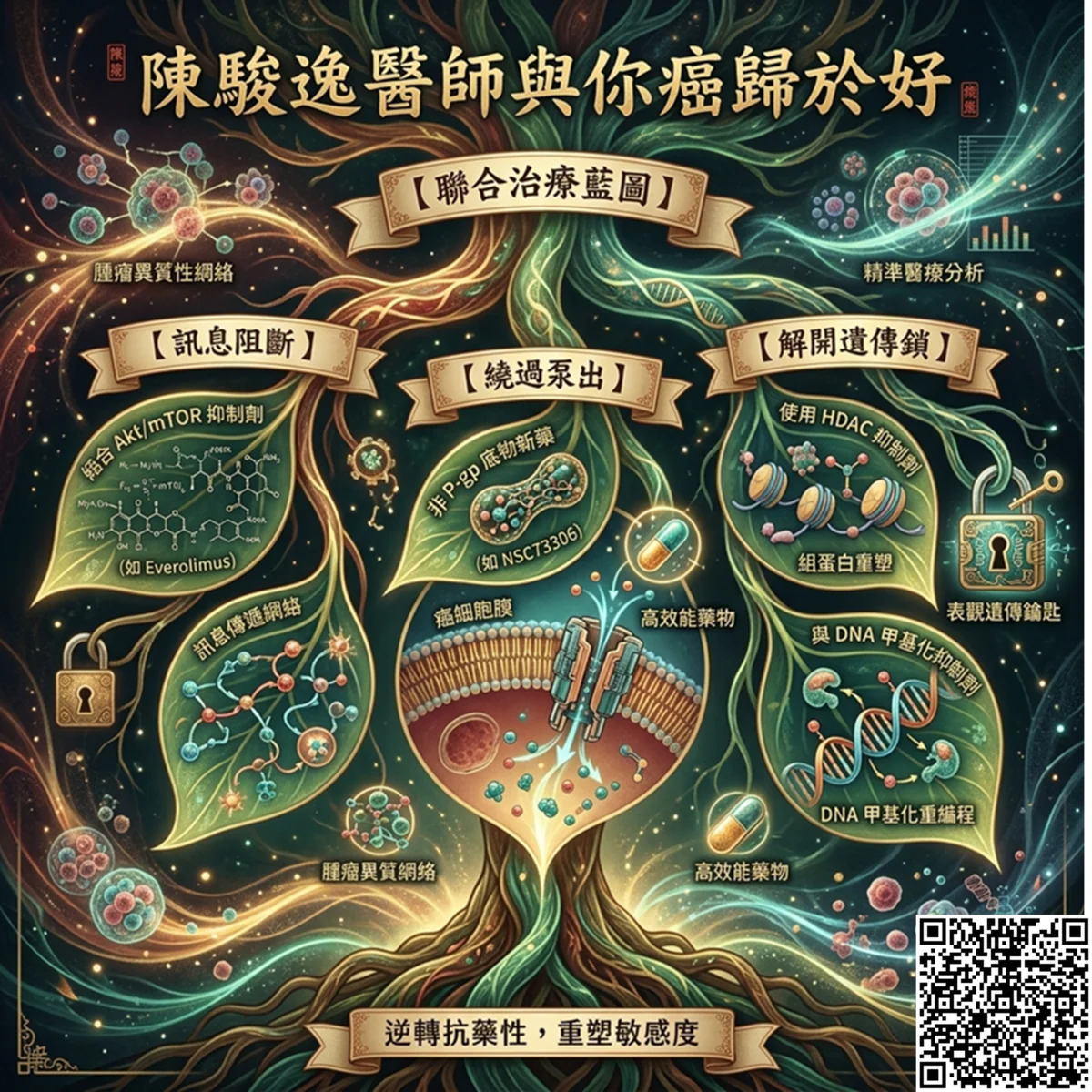
逆轉抗藥性:精準協同的解決方案
透過標靶 PI3K/Akt/mTORC1 軸心、研發不是 P-gp 底物的藥物,並結合表觀遺傳抑制劑,可望突破臨床抗藥僵局。要重新拉住這輛失控的賽車,臨床上的解決方案必須採取「多管齊下」的協同策略:
- 阻斷核心軸心: 目前多項臨床試驗正全面圍剿 PI3K/Akt/mTORC1 訊息軸。例如,使用 mTOR 抑制劑(如 Everolimus)聯合內分泌療法或 Trastuzumab,或使用泛 PI3K 抑制劑(如 BKM120)聯合化療,皆在逐步實證能重新喚醒腫瘤對一線藥物的敏感度。
- 繞過與利用外排泵: 針對 P-gp 過度表現引發的化療抗藥性,除研發不作為 P-gp 底物的新型微管穩定劑外,亦有策略嘗試利用如 NSC73306 等化合物,使其細胞毒性與 P-gp 表現量成正相關,實施反向殲滅。
- 微調核酸與解開表觀鎖: 反義核酸技術(Antagonist)正針對 miR-21 與 miR-221 進行標靶開發。同時,組蛋白去乙酰化酶抑制劑(HDAC Inhibitors,如 :Panobinostat)與 DNA 甲基化抑制劑的臨床試驗亦在開展中,期盼能重塑染色質構型,解開抑癌基因的靜默狀態。

精準剖析趨同抗藥分子網絡,並善用聯合生物標靶調控,是帶領乳腺癌晚期個體化治療走向下一里程碑的關鍵。
面對多藥抗藥性乳癌雖然是一場複雜的臨床硬仗,但當我們將視野放大到整體細胞訊息與表觀遺傳學的趨同網絡時,就會發現各亞型之癌細胞背後共用的生存藍圖。透過精準剖析 PI3K/Akt 軸心失衡、miRNA 靜默與表觀遺傳學密碼,我們正在把過去單一、易被繞過的直線標靶,升級為立體編織的捕獸網。
未來,隨著更多結合訊息阻斷劑與表觀遺傳微調藥物的臨床數據出爐,我們將更有信心為晚期復發的患者,重塑對治療的敏感度,實現真正的個體化精準抗癌。
參考文獻:
Multidrug-resistant breast cancer: current perspectives
Breast Cancer (Dove Med Press). 2014 Jan 10:6:1-13.
#陳駿逸醫師與你癌歸於好
#陳駿逸醫師
#與你癌歸於好
#乳癌治療精準醫學
#抗藥性機制
#乳癌抗藥性
#MDR
#PI3KAkt路徑
#mTORC1
#PTEN
#抑癌蛋白
#microRNA
#miR21
#表觀遺傳學
#DNA甲基化
#HDAC
#Pgp外排泵
#HDAC抑制劑
#DNA甲基化抑制劑
#雌激素受體
#ERα
#ERβ
#BRCA1
#表觀遺傳修飾
#組蛋白乙醯化
#甲基化
#HDAC
#DNMTs
#CpG島超甲基化
#非編碼 RNA
#miRNA-27a
#miR-148a
#circRNA
#circGFRA1
#陳駿逸醫師
#台中市全方位癌症關懷協會
陳駿逸醫師醫療門診服務時段
https://mycancerfree.com/contact/
更多陳駿逸醫師的癌症衛教影片請連接https://www.youtube.com/@mycancerfree
更多腫瘤治療相關資訊 請連接”陳駿逸醫師 與你癌歸於好” https://mycancerfree.com