血液腫瘤科/細胞治療中心/台灣細胞免疫醫學會 陳駿逸醫師
學革命
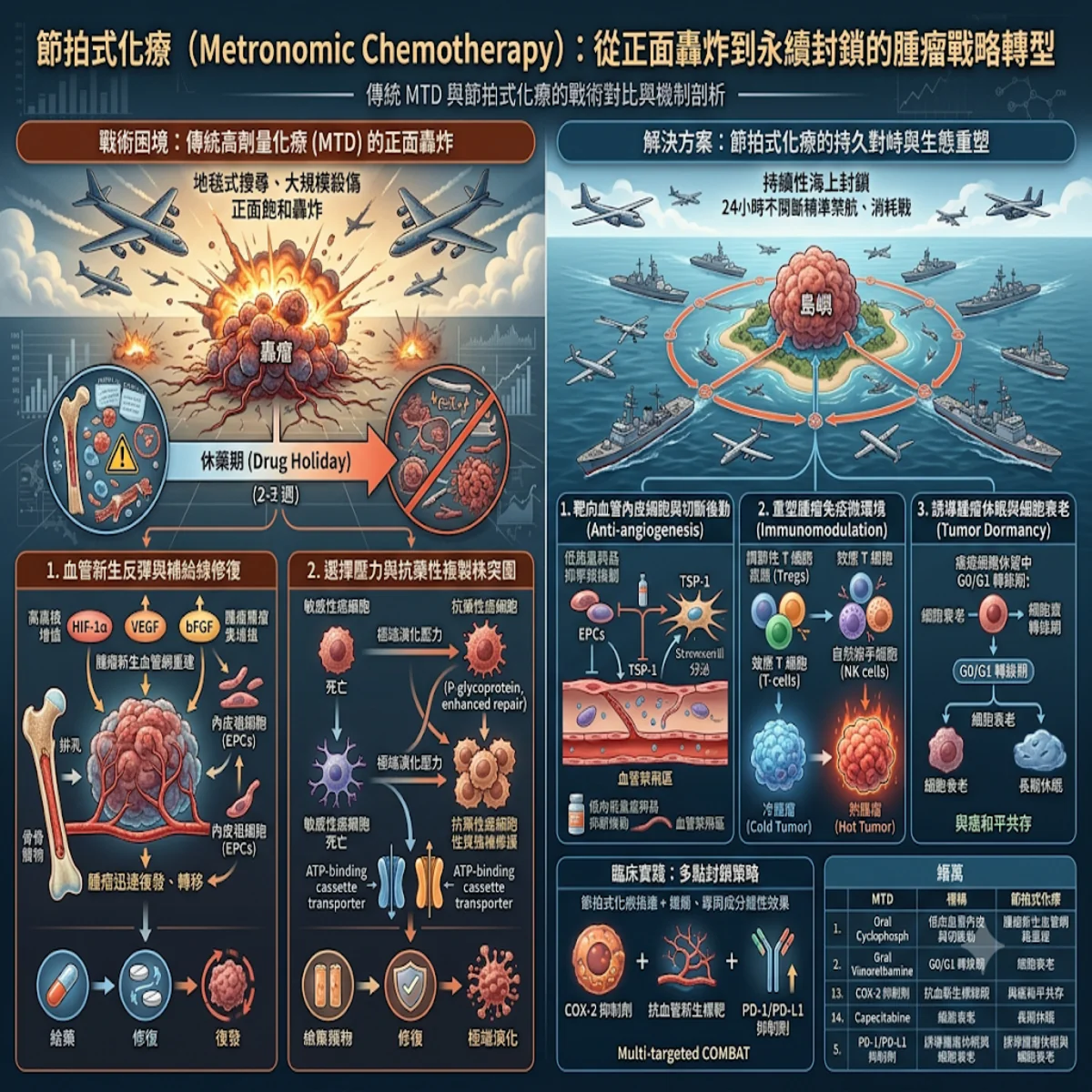
- 問題背景:基因體學之外的隱形高牆——乳癌的抗藥與復發困境
乳癌(在基因體學研究上雖已取得顯著進展,但其高度的分子異質性(Molecular Heterogeneity)仍是臨床治療上的棘手難題。 根據世界衛生組織的最新統計,乳癌已成為全球最常被診斷出的惡性腫瘤之一,更是導致女性癌症相關死亡的主要死因 。臨床上,我們習慣依據雌激素受體(ER)、孕酮受體(PR)及人類表皮生長因子受體2(HER2)的表達譜,將其區分為不同分子雅型 ,並進一步細分為 Luminal A、Luminal B、HER2 富集型、Basal-like 及 Claudin-low 等六種分子亞型 。
傳統的標靶治療、內分泌(荷爾蒙)治療與常規化療雖然初期成效顯著,但在對抗晚期轉移或復發時,高比例的獲得性抗藥(Acquired Resistance)往往令臨床醫師陷入困境。 舉例而言,高達 75% 的乳癌患者為ER alpha 陽性,但在接受抗雌激素(如 Tamoxifen)或芳香環酶抑制劑(AI)治療一段時間後,大約 20% 至 40% 的轉移性患者會因ESR1基因突變(如:常見的 Y537N/C/S 與 D538G 突變)或ESR1基因融合,導致雌激素受體配體結合域(LBD)的缺失,使腫瘤細胞轉化為非配體依賴性的持續活化狀態 。此外,細胞週期調控異常(如: CDK4/6 活性過高、抑制因子 FAT1 功能喪失引發的 YAP/TAZ 活化)以及 PI3K/AKT/mTOR 訊號通路的主動代償,更進一步加劇了抗藥性細胞株的擴增 。
令人困惑的是,許多臨床上發生常規治療失效或早期復發的病例,在進行次世代定序(NGS)時,往往找不到任何明確的基因序列突變。 這種遺傳學結構與臨床表型(Phenotype)之間的巨大落差表明,乳癌的惡性演進與抗藥機制,絕無法單純用「DNA 序列改變」的基因體框架來解釋 。在宿主基因序列完全未變的前提下,腫瘤細胞顯然運用了一套隱形的細胞重編程(Reprogramming)系統,悄悄關閉了腫瘤抑制基因(Tumor Suppressor Genes),並瘋狂放大致癌訊號網絡 。這堵基因體學之外的隱形高牆,正是當前乳癌精準醫療亟待跨越的鴻溝 。
- 特定醫學機制:解構 DNA 甲基化與組蛋白修飾的細胞動態重編程
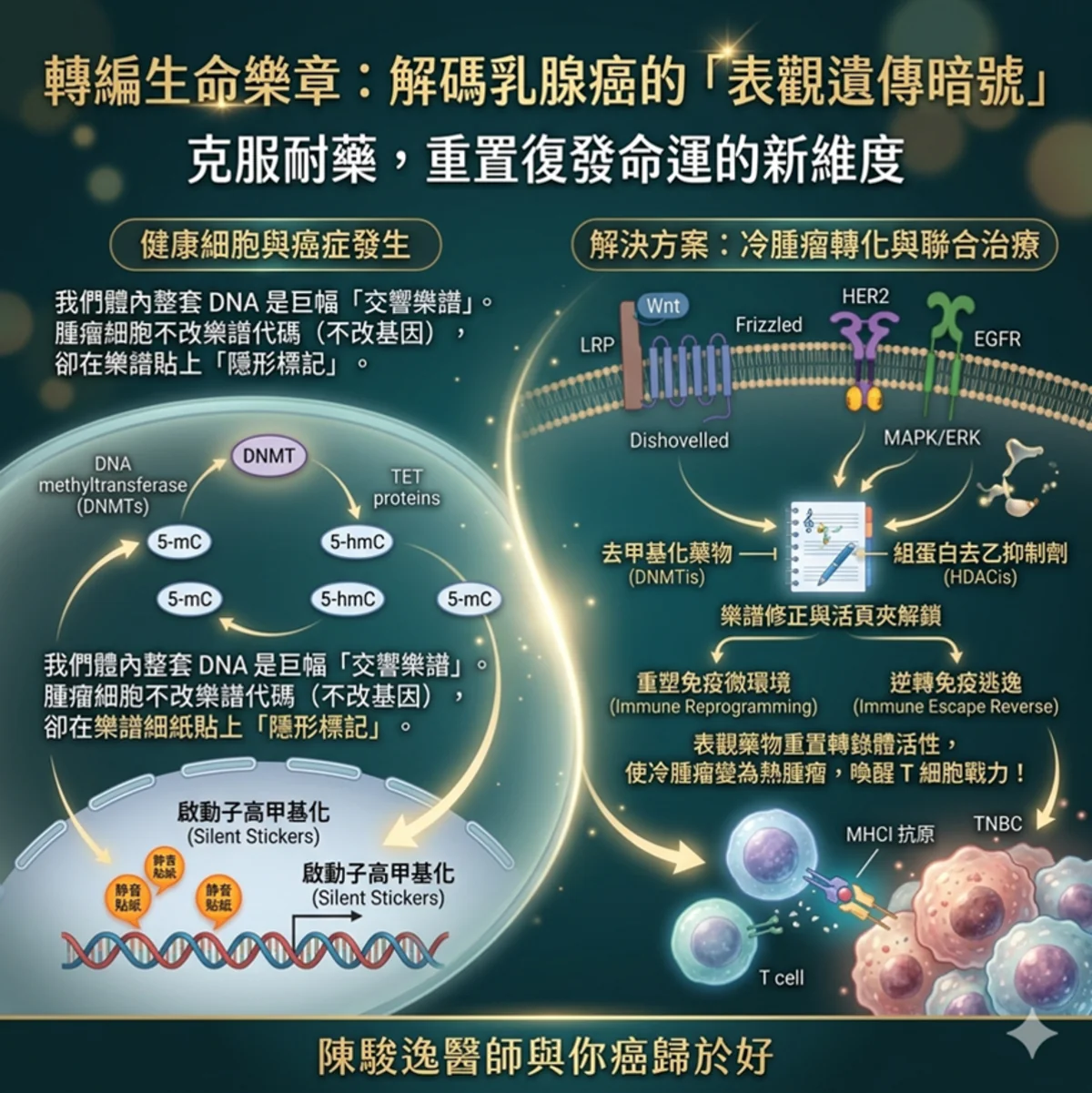
為了透徹理解這套隱形重編程系統,我們必須將目光投向「表觀遺傳學調控(Epigenetic Modifications)」。這是一套在不改變核苷酸序列的前提下,藉由調控染色質結構來決定基因開關的精密機制。 表觀遺傳調控的核心主要由 DNA 甲基化(DNA Methylation)、組蛋白共價修飾(Histone Modifications)以及非編碼核糖核酸(主要是 miRNAs)共同構建而成 。在乳癌病理生理演進中,這些機制交互作用,徹底扭曲了正常的轉錄體學活性(Transcriptomic Profile) 。
為了讓這個微觀的生化過程更具像化,我們不妨將乳癌細胞內整套大約 30 億個鹼基對的 DNA 序列想像成一張宏大而固定的「交響樂譜」。在健康細胞(正常的樂團)中,樂團會嚴格按照譜面上的標記進行演奏,該高昂時高昂(激活腫瘤抑制基因),該靜音時靜音(關閉致癌基因)。然而,當細胞踏上癌變與抗藥的道路時,這張「樂譜」的音符序列(基因序列)雖然沒有被篡改,但細胞卻在樂譜上塗抹了大量的「隱形標記」。
- DNA 甲基化(DNA Methylation)就像是貼在特定音符上的「靜音貼紙」 。當 DNA 甲基轉移酶(DNMTs)在腫瘤抑制基因的啟動子(Promoter)區域瘋狂貼上甲基基團(貼紙)時,負責前來閱讀樂譜並演奏的轉錄因子與 RNA 聚合酶就再也看不到這些音符,導致關鍵的防癌基因(如: BRCA1、PTEN)徹底被「消音」 。反之,在整張樂譜的大範圍背景中(Gene bodies 或全基因體),腫瘤細胞又會大面積地撕掉貼紙(Genome-wide Hypomethylation),使得原本被封印的致癌基因被大聲朗誦,引發染色質極度不穩定與腫瘤演進 。
- 組蛋白修飾(Histone Modification)則如同調整樂譜架空間與演奏力度的「活頁夾與重音記號」。DNA 這根長達兩公尺的琴弦,是緊緊纏繞在一顆顆被稱為「組蛋白(Histone)」的捲軸上的。當組蛋白乙醯化酶(HATs)對組蛋白尾部的離胺酸進行乙醯化(加裝帶負電的乙醯基)時,就像把緊繃的活頁夾鬆開,讓染色質結構變得鬆散(Euchromatin),允許演奏者輕鬆翻閱並大聲朗讀基因;而當組蛋白去乙醯化酶(HDACs)瘋狂工作、拔除乙醯基時,活頁夾會瞬間被鐵夾鎖死,染色質高度凝聚成緊密結構(Heterochromatin),基因被重重包裹,徹底喪失轉錄活性。與此同時,組蛋白甲基轉移酶(如: PRC2 複合物中的 EZH2)則像是在組蛋白特定位點(如: H3K27 或 H3K9)刻上「錯誤的重音記號」,導致特定染色質區段被長期鎖定在非正常的壓制狀態,這正是乳腺細胞躲避抗癌藥物攻擊、維持幹細胞特性(Stemness)的核心暗黑魔術 。
- 解決方案:逆轉遺傳命運的雙重夾擊——表觀遺傳靶向藥物與免疫治療的協同革命
不同於一旦發生便覆水難收的基因突變,表觀遺傳修飾具有高度的「可逆性(Reversibility)」,這為我們打破乳癌抗藥僵局提供了全新的戰略切入點。 現階段,被統稱為「表觀藥物(Epidrugs)」的標靶化學小分子已陸續進入臨床試驗或獲得批准 。這些藥物不再試圖去修改突變的基因,而是直接化身為「貼紙清除劑」與「活頁夾解鎖工具」,強制將耐藥性腫瘤細胞的轉錄體學狀態重置(Reset)回敏感的狀態 。
去甲基化藥物(DNMTis)與組蛋白去乙醯化酶抑制劑(HDACis)的聯合應用,能有效重塑乳癌的免疫微環境,打破腫瘤的免疫逃逸。 在三陰性乳癌等高度惡性的臨床範式中,腫瘤細胞常常藉由表觀遺傳沉默,關閉其干擾素刺激基因(ISGs)與 STING 通路,呈現抗原遞呈缺陷的「冷腫瘤(Cold Tumor)」表型 。當我們引入 HDAC 抑制劑(如: Entinostat 或 Trichostatin A)與 DNMT 抑制劑時,能強制恢復 MICA、ULBP2 等細胞表面標誌物的表達,並誘導腫瘤相關巨噬細胞(TAMs)朝向具備抗腫瘤活性的 M1 型極化 。更重要的是,HDACi 能夠逆轉因長期治療引發的 T 細胞竭竭(T-cell Exhaustion),重新喚醒腫瘤浸潤淋巴細胞(TILs)的細胞毒性殺傷功能 。
【表觀遺傳協同聯合療法作用機制範式】


臨床轉化醫學的數據表明,表觀藥物與免疫檢查點抑制劑的聯合療法(Combined Drug Therapy, CDT),展示出顯著的同盟協同效應。 例如,在針對三陰性乳癌的早期臨床試驗中,將抗 PD-L1 單株抗體 Atezolizumab 與 HDAC 抑制劑 Entinostat 聯合使用,其客觀緩解率(ORR)與無進展生存期(PFS)皆顯著優於單純免疫治療 。此外,針對致癌性組蛋白甲基轉移酶 EZH2 的抑制劑 Tazemetostat,與免疫治療共用時,能顯著減少腫瘤微環境內骨髓衍生抑制細胞(MDSCs)的浸潤,成功將原本對免疫治療冷漠的乳腺癌病灶轉化為對免疫細胞敞開大門的「熱腫瘤(Hot Tumor)」。這種雙重夾擊的策略,正逐步成為臨床克服三陰性乳癌廣泛異質性與多藥抗藥的殺手鐧 。
- 結語:從代碼修改到系統重塑——乳癌精準醫療的下一維度
綜上所述,乳癌的臨床處置已正式跨越了單純依賴「基因體序列定序」的傳統範式,邁向了更動態、更立體的表觀遺傳調控時代。 透過深入解析 DNA 甲基化、組蛋白代償性修飾以及非編碼 RNA 網絡的微觀變革,我們終於得以看清腫瘤細胞在抗藥性演進中那張抹滿「靜音貼紙」與「錯誤重音記號」的暗黑樂譜 。表觀遺傳修飾的可逆特質,無疑為我們點亮了重新編排這首抗癌交響樂的曙光 。
展望未來,將表觀藥物精準融入現有的額爾蒙、標靶及免疫治療框架中,必將成為攻克乳腺癌耐藥屏障的必然趨勢作為常年穿梭於病榻第一線的腫瘤科醫師,我們不僅要學會用手術刀切除病灶、用標靶藥物阻斷特定代碼,更需要學會利用表觀遺傳學工具去「重塑細胞的生存系統」。當我們徹底掌握了這套表觀遺傳密碼的解鎖鑰匙,那些曾經令我們束手無策的晚期耐藥與復發型乳腺癌,終將在多維度的聯合精準打擊下,迎來可防、可控、可逆轉的全新療癒轉機 。

HER2和Wnt/β-catenin訊息傳遞路徑是乳癌細胞進展中兩個至關重要的路徑。 HER2訊息傳遞路徑透過二聚化活化PI3K/AKT等多種其他通路,進而促進EMT遷移,最終導致癌症進展。 TGFβ的功能在於抑制細胞增殖,進而阻止癌症進展。儘管TGFβ可以增強SMAD3 S208位點的磷酸化,但它也會放大PI3K/AKT訊號通路,進而促進乳癌的發展。此外,Wnt/β-catenin路徑也能促進乳癌的發展,其機制是透過β-catenin進入細胞核並影響TCF/LEF複合物,加劇病情。
 乳癌中正常細胞和腫瘤細胞的表觀遺傳學改變。 DNA甲基化和組蛋白修飾等表觀遺傳修飾會改變DNA的可及性和染色質結構,進而調控基因表現模式。在正常細胞中,DNA甲基轉移酶(DNMTs)在胞嘧啶嘧啶環的5位添加甲基,此過程稱為DNA甲基化。 Ten Eleven Translocation (TET)蛋白家族催化5-甲基胞嘧啶(5mC)氧化為5-羥甲基胞嘧啶(5hmC)、5-甲醯胞嘧啶(5fC)和5-羧基胞嘧啶(5caC)。因此,TET蛋白提供了一條主動的DNA去甲基化途徑,並與基因表現調控密切相關。 TET蛋白也可以透過胸腺嘧啶DNA糖苷酶(TDG)切除5fC和5caC來介導主動去甲基化。此外,組蛋白乙醯轉移酶(HATs)透過將乙醯輔酶A上的乙醯基轉移至組蛋白上保守的離胺酸殘基,形成ε-N-乙醯賴氨酸,從而乙醯化這些離胺酸殘基。同時,組蛋白H3第27位賴氨酸的甲基化(H3K27me1/2/3)由染色質修飾酶-多梳抑制複合物2(PRC2)催化。 PRC2能夠維持基因轉錄抑制和正常的生物體發育。然而,癌細胞具有不同的表觀遺傳特徵。癌細胞的表觀遺傳特徵中甲基化程度降低。這種甲基化水平的降低會影響大量基因的活性。由於甲基化與基因活性降低有關,低甲基化可能增強受影響基因的活性。在這種情況下,如果相關基因參與細胞生長,可能導致細胞分裂,從而促進癌症進展。此外,在不同類型的癌細胞(例如乳癌細胞)中均發現了組蛋白去乙醯化酶(HDAC)活性增強,其特徵是組蛋白乙醯化標記的缺失。組蛋白甲基轉移酶(HMT)是為組蛋白添加甲基的酶,而組蛋白去甲基化酶(HDM)則具有相反的功能。在乳癌細胞中,HMT可能會改變,導致甲基被錯誤地放置在錯誤的位置,從而抑制腫瘤抑制基因的表達。所有這些事件以及SWI/SNF染色質重塑複合物亞基的突變都可能導致染色質結構異常,從而促進癌症的發生和發展。
乳癌中正常細胞和腫瘤細胞的表觀遺傳學改變。 DNA甲基化和組蛋白修飾等表觀遺傳修飾會改變DNA的可及性和染色質結構,進而調控基因表現模式。在正常細胞中,DNA甲基轉移酶(DNMTs)在胞嘧啶嘧啶環的5位添加甲基,此過程稱為DNA甲基化。 Ten Eleven Translocation (TET)蛋白家族催化5-甲基胞嘧啶(5mC)氧化為5-羥甲基胞嘧啶(5hmC)、5-甲醯胞嘧啶(5fC)和5-羧基胞嘧啶(5caC)。因此,TET蛋白提供了一條主動的DNA去甲基化途徑,並與基因表現調控密切相關。 TET蛋白也可以透過胸腺嘧啶DNA糖苷酶(TDG)切除5fC和5caC來介導主動去甲基化。此外,組蛋白乙醯轉移酶(HATs)透過將乙醯輔酶A上的乙醯基轉移至組蛋白上保守的離胺酸殘基,形成ε-N-乙醯賴氨酸,從而乙醯化這些離胺酸殘基。同時,組蛋白H3第27位賴氨酸的甲基化(H3K27me1/2/3)由染色質修飾酶-多梳抑制複合物2(PRC2)催化。 PRC2能夠維持基因轉錄抑制和正常的生物體發育。然而,癌細胞具有不同的表觀遺傳特徵。癌細胞的表觀遺傳特徵中甲基化程度降低。這種甲基化水平的降低會影響大量基因的活性。由於甲基化與基因活性降低有關,低甲基化可能增強受影響基因的活性。在這種情況下,如果相關基因參與細胞生長,可能導致細胞分裂,從而促進癌症進展。此外,在不同類型的癌細胞(例如乳癌細胞)中均發現了組蛋白去乙醯化酶(HDAC)活性增強,其特徵是組蛋白乙醯化標記的缺失。組蛋白甲基轉移酶(HMT)是為組蛋白添加甲基的酶,而組蛋白去甲基化酶(HDM)則具有相反的功能。在乳癌細胞中,HMT可能會改變,導致甲基被錯誤地放置在錯誤的位置,從而抑制腫瘤抑制基因的表達。所有這些事件以及SWI/SNF染色質重塑複合物亞基的突變都可能導致染色質結構異常,從而促進癌症的發生和發展。
文獻引用來源 (References):
- Karami Fath M, et al. Cellular & Molecular Biology Letters (2022) 27:52. (The role of epigenetic modifications in drug resistance and treatment of breast cancer)
#陳駿逸醫師與你癌歸於好
#表觀遺傳學
#乳癌
#表觀藥物
#聯合免疫治療
#重塑免疫微環境
#乳癌精準醫療 #表觀遺傳學 #液態切片 #奈米醫學 #轉化醫學 #抗藥性逆轉 #陳駿逸醫師與你癌歸於好#翻轉基因宿命
#乳癌治療
#精準醫療
#個體化醫學
#Personalized Medicine
#表觀遺傳學
#Epigenetics
#DNA甲基化
#高甲基化
#低甲基化
#組蛋白修飾
#緊密染色質
#鬆散染色質
#Euchromatin
#非編碼RNA
#乳癌抗藥性
#腫瘤異質性
#癌症幹細胞
#三陰性乳癌
#免疫逃逸
#表觀遺傳藥物
#Epi-drugs,利用可逆性重新喚醒抑癌基因)
#DNMTi
#HDACi
#聯合治療
#免疫檢查點抑制劑
#冷腫瘤轉熱
#液態切片
#ctDNA
#微小殘留病灶
更多腫瘤治療相關資訊 請連接”陳駿逸醫師與你 癌歸於好” https://mycancerfree.com
更多癌症病友需知 請連接”全方位癌症關懷協會” https://www.cancerinfotw.org/index.php
歡迎參與臉書社團:陳駿逸醫師的用心話聊俱樂部 www.facebook.com/groups/456281992960876/
陳駿逸醫師門診的服務資訊
https://mycancerfree.com/contact/